分析人员必读!片剂溶出度相关知识汇总!!
溶出度(Dissolution rate)也称溶出速率,是指在规定的溶剂和条件下,药物从片剂、胶囊剂、颗粒剂等固体制剂中溶出的速度和程度。测定固体制剂溶出度的过程称为溶出度试验(Dissolution test),它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法。药物溶出度检查是评价制剂品质和工艺水平的一种有效手段,可以在一定程度上反映主药的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的差异;在产品发生某些变更后(如处方、生产工艺、生产场所变更和生产工艺放大),确认药品质量和疗效的一致性;也是评价制剂活性成分生物利用度和制剂均匀度 的一种有效标准,能有效区分同一种药物生物利用度的差异,因此是药品质量控制必检项目之一。
一般认为,难溶性 (一般指在水中微溶或不溶) 药物,因制剂处方与生产工艺造成临床疗效不稳定的药物以及治疗量与中毒量相接近的药物(包括易溶性药物),其口服固体制剂质量标准中必须设定溶出度检查项。另外固体制剂的处方筛选及生产工艺流程制订过程中,也需对所开发剂型的溶出度做全面考察。一个可行的溶出度试验法应是在不同时间、地点对同一制剂的溶出度测定或不同的操作者之间的测定都必须达到试验结果具有良好的重现性。为了达到以上目的,必须对溶出度测定试验进行 全面充分的研究。
生物药剂学(BCS)分类(美国FDA ):
第1类:高溶解度一高渗透性
第2类:低溶解度一高渗透性
第3类:高溶解度一低渗透性
第4类:低溶解度一低渗透性
高溶解度:单个制剂能在250mL,pH值1.0~8.0介质中溶解——相当于中国药典的“微溶”
高渗透性:绝对生物利用度≥90%
上述分类可以作为设定体外溶出度质量标准的依据,也可用于预测能否建立良好的体内-体外相关性的依据。BSC提示,对于高溶解度、高渗透性(1类)药物及某些情况下的高溶解度、低渗透性(3类)药物,其溶出度在0.1NHCL中15min时为85%即可保证药物的生物利用度不受溶出的限制,即制剂的行为与溶液相似。
溶出度研究试验主要包括以下内容:(1)溶出介质的选择,(2)溶出介质体积的选择,(3)溶出方法(转篮法与桨法)的选择,(4)转速的选择,(5)溶出度测定方法的验证,(6)溶出度均一性试验(批内),(7)重现性试验(批间)等。
近来在新药审评中发现,部分研究单位在进行溶出度研究时存在一些问题,主要表现在溶出度研究资料过于简单或溶出度研究内容不够全面。现予以具体分析,希望能对溶出度研究有一定的帮助。
1、溶出介质的选择:
介质种类:
水——最常用,良好脱气水的pH值为5.0~7.0, 适用于溶解度对pH值不敏感的药物
盐酸溶液——模拟酸性胃液的介质, 适用于酸中稳定的药物,浓度一般为0.1~0.01mol/L,pH值一般为1.0~2.2
磷酸盐缓冲液(PBS)——模拟中等酸性至弱碱性胃液或肠液的介质, 浓度一般为0.05mol/L,pH值一般为4.5~7.6
醋酸盐缓冲液——模拟中等酸性胃液的介质, 浓度一般为0.05mol/L,pH值一般为3.4~6.0, 醋酸易挥发,导致pH值变化,溶出速率变化、测定时间长的药物不宜采用醋酸盐缓冲液作介质
普通制剂 :
(1)酸性药物制剂 pH值分别为1.0或1.2、5.5~6.5、6.8~7.5和水;
(2)中性或碱性药物/包衣制剂 pH值分别为1.0或1.2、3.0~5.0、6.8和水;
(3)难溶性药物制剂 pH值分别为1.0或1.2、4.0~4.5、6.8和水;
(4)肠溶制剂 pH值分别为1.0或1.2、6.0、6.8和水;
调释制剂 :
pH值分别为1.0或1.2、3.0~5.0、6.8~7.5和水查询药物的PKa值,PKa<3.0,为酸性物质;PKa≥3.0为碱性/中性药物
介质的pH值:
原则:根据体内吸收部位的pH值环境
药物溶解度对pH值的敏感性(pH-溶解度曲线测定:取过量的原料药,置具塞试管中,分别加PH1.0-8.0的溶出介质适量,使之成饱和溶液,过滤,取续滤液经HPLC法测定准确溶度,计算溶解度并绘制pH-溶解度曲线;观察曲线与PH的变化情况)。
药物的稳定性来选择介质的pH值(确保主成分在溶出介质中的稳定性)。
2. 溶出体积的选择
篮法和桨法的溶出体积应在500~1000ml,目前多采用大杯法900-1000ml。除有充足理由。不建议采用该范围以外的体积。该体积范围的设定是为满足药物的漏槽条件 —— 溶出介质体积应大于药物全部溶解所需体积的三倍,以保证药物溶出不受其溶解性影响而设。而对于小杯法是相对小规格固体制剂,当采用紫外法检测无法满足测定要求时,从第二法衍生改良而得,截至目前仅我国收载。但该法对于某些活性成分,由于无法满足溶出度试验漏槽条件的要求,故建议在新产品溶出度研究与拟定上尽量勿采用该法,解决的办法:
①加大进样量浓度至100~200μl。
②调整流动相使主成分保留时间缩短,色谱峰成“尖峰”状,积分将更加准确、精密度自然提高。
③不选最大吸收波长,选择末端强吸收处波长作为检测波长,以加大响应值。
3.溶出方法装置和转速的选择
篮法和桨法是目前通用性最强、耐用性最高、仪器最为普及的法定溶出方法,因此,建议首选该两法,桨法—首选,只要药物不上浮就应选桨法,推荐50转/分,一般选用50~75rpm,不推荐采用100转甚至更高的转速,因为这将严重降低对于不同来源同一制剂的区分力,且大大降低体内外相关性,除非在已验证该制剂体内具有良好生物利用度的前提下,并应提供充足的理由与验证资料;转篮法推荐100转/分,一般选用50~100rpm;桨法通常将转篮法/100转强度看作与桨板法/50转相当、且将该强度看作与中老年人体的胃肠道蠕动强度等同;当采用桨法/75rpm试验条件难以准确评价质量时,可改为篮法/120rpm。但是在某溶出介质中,当结束时间点溶出量仍达不到90%,缓/控85%时,可放宽溶出参数,如现增加转速75转/分,未果的话可以加入表面活性剂。
4. 测定时间点和结束时间点的设定
测定时间点:
普通和肠溶制剂可分别为5、10、15、20、30、45、60、90120min,此后每隔1小时测定
缓/控制剂:15、30、45、60、90分钟和2、3、4、5、6、8、10、12、24小时
结束时间点:
在酸性介质(ph1.0-3.0)中最长为2h,其他PH介质中,普通和肠溶为6h,缓/控为24h,担当连续两点达90%(缓/控85%),且差值小于5%时,可提前结束。
5. 取样时间点和限度的拟定
高溶解度且快速溶解产品:单个时间点,大多数情况。对于普通制剂,建议以第一次出现溶出量均在85%(或90%)以上的两时间点,且该两点溶出量差值在5%以内,取前一时间点作为质量标准中的取样时间点,并将该点的溶出量减去15%作为溶出限度。取样时间一般为5的倍数,3以上可向上约靠。
低溶解度且溶出缓慢的制剂或对溶出度有特殊要求的制剂:两个时间点
缓释制剂:药物作用8小时的一般至少设3个时间点
作用12~16小时的至少设4个时间点
作用24小时或以上的至少设5个时间点
6. 含量测定方法的选择与验证
原则:专属好、重复性好、方便快捷、易于自动化尽可能与含量测定方法相同
紫外分光光度法——首选
无紫外吸收的,可衍生化后比色
溶出度测定普遍采用方便快捷的UV法,但一定要注意辅料的干扰。特别是在短波长处,更应注意辅料的末端吸收等干扰因素的存在。一般认为辅料的干扰应在2%以内,否则UV法不适用。
采用UV法测定前,一定要进行紫外扫描,以考察辅料是否存在干扰。方法为:分别取对照品溶液和供试品溶液(浓度最好与对照品溶液一致或略低),进行紫外扫描。着重观察供试品溶液图谱情况:是否存在基线被抬升的现象,以及与对照品溶液图谱重合的情况。如辅料无干扰,则供试品溶液图谱应与对照品溶液图谱基本重合或整体略低于对照品图谱;如辅料有干扰,就会出现基线或末端吸收处被抬高的现象。
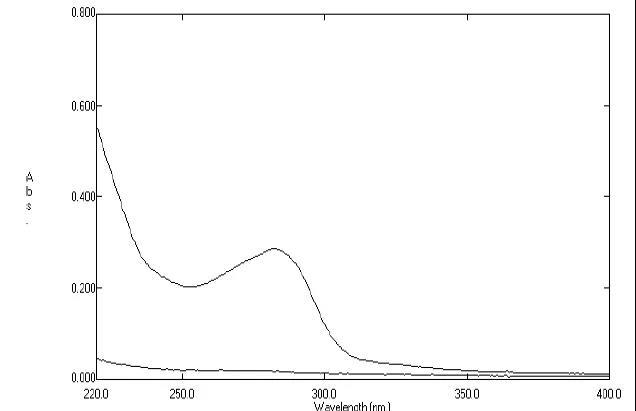
图1 供试品溶液图谱(上方)与对照品溶液图谱(下方)相比,基线被整体“抬高”
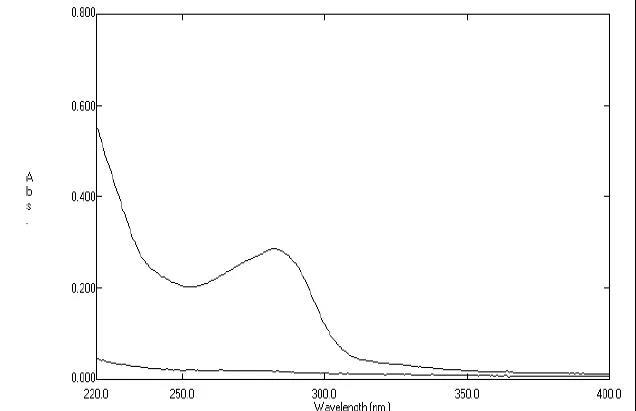
图2 辅料呈一“斜坡形”吸收(下方图谱), 在测定波长处的吸收度值约占供试品溶液(上方图谱)的5%左右
另一方法可采用HPLC法,将其测定结果与UV法进行比较,以判定辅料是否有干扰。
当辅料干扰紫外法测定时,通常可采用以下方法:
双波长吸收度差值法、导数光谱法
HPLC法:在HPLC 上可轻松将辅料与主成分分离,该法测定最为准确、可靠,只是工作量略显增加。
验证:
溶出度检测方法按照既定的方法学认证各项要求验证,尚需注意以下各事项:
(1) 对于某些难溶性药物,如难以采用溶出介质直接溶解对照品,可先加少量有机溶剂(如甲醇或乙醇)溶解后,再加溶出介质稀释的办法,建议最终溶液中有机相比例不超过5%。如超出,应予以相应验证。
(2) 线性范围应考虑到有可能出现的低浓度点情况,如对于溶出曲线或调释制剂的测定等。并为减小外标一点法的测定误差,建议将对照品溶液配制为约50%释放量浓度。
(3) 应注意考察活性成分在37℃溶出介质中的稳定性。若不佳,应在质量标准中标注“取出后立即测定的”字样;不建议在溶出介质中加入抗氧剂、稳定剂等物质。
(4) 根据实际情况,检测波长可选取非最大吸收波长。
(5) 当测定法为紫外法时,由于易受辅料或胶囊壳的影响,建议分别取对照品溶液与供试品溶液进行紫外扫描后根据所得图谱予以明了,亦可采用高效液相法进行佐证。如干扰存 在,可考虑采用双波长相减法或空白胶囊壳扣除法等方法予以消除,不建议采用紫外导数光谱法;如干扰排除较为困难,建议采用高效液相色谱法测定。
(6) 空白胶囊壳干扰在2%以内可忽略不计;若大于2%则应校正,并在该品种项下予以注明;大于25%时,则被视为溶出试验无效,应重新选择测定方法。关于其测定法,建议取6粒样品,彻底清除其中内容物,同法试验和过滤后,分别量取相同体积混匀测定。测定时、以相应溶剂为空白。
(7) 不应出现溶出度测定结果均值高于含量测定结果3%以上的情况。如出现,应重新考察溶出度测定方法的适用性。
(8) 对于小规格制剂,不建议采用大于1cm的吸收池进行紫外法测定,而建议采用HPLC法(并可酌情加大进样量以提高测定精密度)。但对于具有较强紫外吸收的活性药物,为提高工作效率,可在溶出曲线大批量样品测定时,采用0.1cm~1.0cm短吸收池,但必须经过验证。
(9) 对一些小规格制剂,样品过滤时会发生吸附,判定吸附与否的方法可采用:
①取溶出液,分别过滤不同体积的初滤液后测定,观察响应值的变化情况,
②取样后一份不过滤,直接采用高速离心,取上清液测定。并与采用过滤法所得的续滤液比较,考察两者间测定数据的差异。判定自动取样溶出仪的固有滤膜是否存在吸附时,一般采用该法。
③取对照品溶液,经滤膜过滤后,与原溶液进行比较,观察测定前后数据的变化情况。
如滤膜对药物有吸附,可采用将滤膜在沸水中煮沸1h,或加大初滤液体积等办法予以解决。但建议采用直接高速离心的方法。
7. 溶出曲线比较
产品上市发生较小的变更时,采用单点溶出度试验可能就足以确认其是否未改变产品的质量和性能。发生较大变更时,则推荐对变更前后产品在相同的溶出条件下进行溶出曲线比较。在整体溶出曲线相似以及每一采样时间点溶出度相似时,可认为二者溶出行为相似。
由于多pH值溶出曲线的绘制已成为剖析和表达固体制剂内在品质的重要手段,故对溶出曲线比较的科学评价愈发重要。截至目前,报道有多种比较方法。
但自美国和日本等国的官方机构认定采用模型非依赖方法之一的“相似因子比较法”之后,现基本上被统一采纳。该法特点是对溶出曲线进行整体评价,通过计算相似因子(ƒ2)比较溶出行为的相似性。
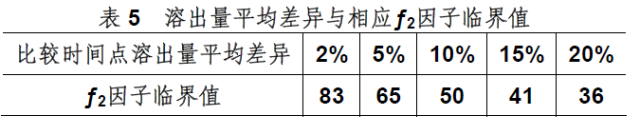
通常,当ƒ2数值介于50-100认为两条溶出曲线是相似的,该数值限定是基于两条比较曲线上任一比较时间点溶出量平均差异限度不大于10%的考虑。表5提供了溶出量平均差异与相应ƒ2因子临界值的关系。
对于普通制剂,15min时溶出量达85%以上,则无需进行曲线比较,此时仿制制剂亦满足此条件。
展源
何发
相关文章
-

生物药剂学分类系统与溶出度试验
2020-05-27
-

生物药剂学分类系统与溶出度试验的关系
2020-05-27
-

化学药品溶出度方法研究
2020-05-27
-

溶出度技术的应用
2020-05-27
-

关于溶出度技术的应用
2020-05-27
-

溶出度试验质量标准制订
2020-05-27
-

化学药品溶出度方法实验研究初探
2020-05-27
-

溶出度实验及测定方法验证
2020-05-27
-

溶出度实验、测定方法验证
2020-05-27
-

溶出度试验质量标准的制订
2020-05-27

加载更多