很多人在从事药学
相关工作的时候,都希望有一份经验宝典能让他们更快的适应以及面对常见问题时能尽快应对,随小析姐一起来看看药学
里面关于
的使用TIPS吧。
01有关中药
在做中药材的浸出物的检测时,一定要按标准控制好溶剂的浓度(如乙醇等),否则检验结果会差异很大。
中药制剂的溶液(比较稠或量少)要用滤纸过滤时,可以先通过离心的方法使之分层,再去过滤。这样可以加快过滤,减少有机溶剂的挥发(环境温度高时).
在做人参皂苷RB1和黄芪甲苷时,如果同
中酸的浓度很高,这两个成分都会分解,从而测不出含量,所以应避免和挥酸时同时进行含量前处理。
中药注射剂检验树脂项时,用的氯仿最好用优级的,不同的试剂对结果影响很大。同时在提取放置的时间尽量长些,使其完全分开。
用高效液相色谱仪检测人参、麻黄的含量时因检测波长为边缘波长(203、207nm)往往一次不能成功,特别是使用一段时间后的检测器。可卸掉
,直接连接检测器,用0.1%的盐酸清洗检测池4小时,效果会好些。
中药液相测含量时,因每次配制的流动相有差异,按标准配制的流动相,有时峰分不开,可以在药典允许的范围内适当调整比例,改善效果。
在温度低的环境下做一些中药制剂的萃取时,往往会出现乳化现象,即两相不容易分层,这时可加入少量的饱和氯化钠溶液或者用电吹风对分液漏斗加温或用热抹布敷,可以加速其分层。
当液相鉴别中供试品与对照品出峰时间不一致,无法判断是否合格时,可用对照品与供试品各半配成混合溶液后进样,若峰宽未变宽,未出现驼峰,即可判断为合格。
做原料残留物检测的时候,如果主药对杂质有干扰,现有方法无法检出,需要自己建方法的话,要优先考虑利用理化性质将杂质分离出来再进行测定。往往有意想不到的效果。
在采用HPLC法测物质含量时,如若流动相中用了缓冲盐,一定要注意其pH值,放置过程中可能会产生变化,而某些药物对这种变化很敏感。
用HPLC法测物质含量时,温度的控制极其重要,最好用有柱温箱的,如果没有就要装空调保持恒温后再测定,否则会出现基线飘移,结果不准确。
有些品种温度的影响非常大,遇到一个品种,对照品溶液不能放在冰箱中,放在液相室内还开着空调,第二天才能做,否则第二天的对照品到中午也不能用。
用HPLC法测物质含量时,样品最好用流动相溶解,否则容易产生干扰峰,影响结果,特别有可能出现倒峰,一定要注意。使用含有缓冲盐的流动相,容易堵塞管路和柱子,甚至检测器,如果检测器堵了,最好先不要拆,使用10%甲醇水超声加热一下作为流动相,慢慢小流量冲洗,效果很明显。
用高效液相色谱仪测定含量时,用色谱纯试剂处理对照品和样品,可减少误差。
HPLC检测中,梯度条件容易产生干扰峰,可尝试通过以下几种方法规避:
1、使用重蒸后的水或用市售的纯净水(娃哈哈的比较好)
在做片剂或胶囊剂的溶出度实验时,规定药液需经0.8Um的滤膜滤过,但采用液相检测时,进柱前样品还要经0.45Um的滤膜,此时,可以省略第一步过滤,直接做进柱前的样品过滤,否则滤膜会对样品产生吸附,使检测结果偏低。另外不同生产厂家的滤膜对样品的吸附不同,在检测时一定要要注意,可用对照品先做一个过滤前后的对比试验,检查滤膜的吸附。
使用HPLC的过滤装置时 ,一定要注意虑膜的材质,如果是“羟甲基纤维素”不可以过滤含有机相的液体,否则就溶解了,有机相过滤要使用聚四氟乙烯的。 滤膜材质与溶剂的兼容性如下表。
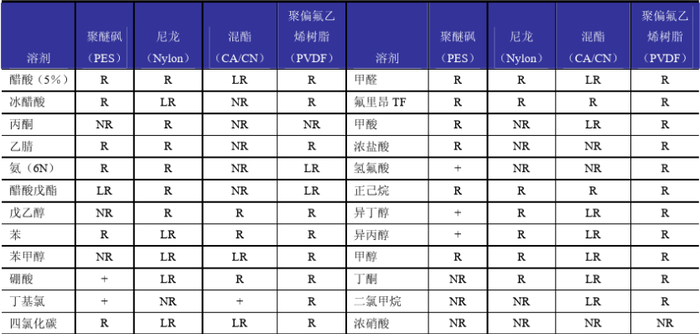
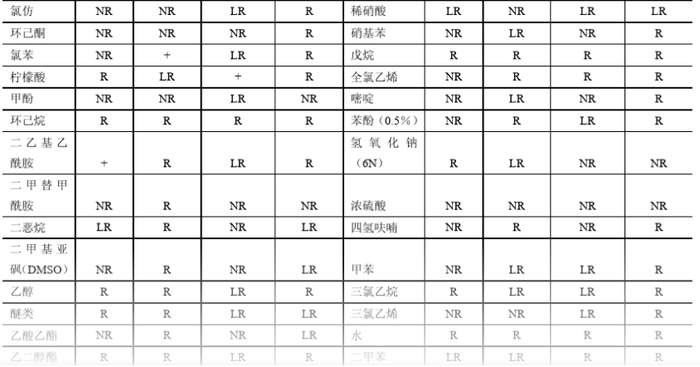
当做高效液相测定肽类时,使用梯度条件情况下,在做第一针样品前,最好走一个空梯度,这样保留时间会一致些。
在用HPLC做定量检测时,柱温和流动相的比例要尽量保持一致,否则,保留时间和积分面积会发生变化,影响最终的检测结果。
在做HPLC时,假如发生重叠峰的现象,通常可以尝试以下几种方法:
高浓度的NaOH标准溶液(比如0.5M)在使用过程中,桶底的部分往往会浓度变高,一般十升的溶液,最下面的一升就不能用了,否则会给滴定结果带来很大的偏差,尤其是夏天。
当液相鉴别中供试品与对照品出峰时间不一致,无法判断是否合格时,可用对照品与供试品各半配成混合溶液后进样,若峰宽未变宽,未出现驼峰,即可判断为合格。
用HPLC法测定酚酸等不稳定成分时,可将对照品溶液及供试品溶液调成酸性(可用磷酸等),这样即不会影响测定结果,而且样品也比较稳定,保存时间比较长。PH值一般调到2左右即可!
在进行HPLC测定时,所用的水最好是双蒸水,这样对测定结果干扰少,而且要勤换,如果通道生霉,可用异丙醇冲洗通道.有机相如已腈最好滤一下,即使是进口色谱纯溶剂。
1、使用预柱保护分析柱(硅胶在极性流动相/离子性流动相中有一定的溶解度);
2、大多数硅胶基质反相
的pH稳定范围是2-8,尽量不超过该
的pH范围;
5、如果使用极性或离子性的缓冲溶液作流动相,应在实验完毕柱子冲洗干净,并保存于甲醇或乙腈中;
6、氯化物的溶剂对其有一定的腐蚀性,故使用时要注意,柱及连接管内不能长时间存留此类溶剂,以避免腐蚀。
用HPLC法测物质含量时,温度的控制是很重要,最好用有柱温箱的,如果没有就要装空调保持恒温后再测定。但不至于一定出现基线飘移,更多时候是保留时间的变化,对结果影响也不一定,有时不会影响准确度。
在做分析方法验证时,鉴别项的专属性一般都很强,像红外、紫外等,可不做专属性,但是注意显色反应的空白溶液,要保证溶液本身不显色。
如果做过三硅三酸镁的检测,是否会遇到这样一个问题:重金属检测时按照标准进行到最后,得到的样品呈现红色,与对照品的颜色(黄棕色)无法进行对照,其主要原是因为在此标准中加入酚酞调节溶液的酸碱度,最后一步加入硫代已酰胺试液后,溶液显碱性,使酚酞显红色。解决的方法是在最后加入盐酸进稍微多加一点,使溶液显酸性,最终使对照与样品颜色一致。
在做比色法测定环氧乙烷残留时。若加入品红后等待1h后不见比色管(实验中加入已二醇体积>0.8ml的比色管)呈微红色,则证明实验中所使用的高碘酸失效,需重新配置高碘酸。
在含量测定过程中,如果遇到测量结果不准时如何处理?根据经验在测量过程中可以带标准样品(已知含量)和被测试样品同时、平行进行测试。把测试结果和标准样品进行比较即可。这样的话,测试的结果就可以得到修正了。如已知含量的标准样浓度为1.0,而测试结果为0.91,就需要把样品的测试结果除以0.91即可得到相对准确的结果。采用“加标回收”的方法更好。只是相对复杂一些。
HPLC测含量时流动相若是乙腈,乙腈最好是新打开的,使用放置时间过长的乙腈容易导致检测不出主峰。
在用HPLC测定肝苏胶囊的含量时,其盐酸的浓度相当重要,浓度一定要够才行哟。
在做高液时,最好一次把流动相配足但要注意流动相的有效期,如果先配的流动相不够,再用相同的方法去配的流动相继续用,会出现保留时间不一致.其次,在发现流动相不够时,最好不要把流出来的"废液"再收集起来继续用,这样出来的峰面积会增大的。
HPLC流动相用到盐的时候,定期用纯水清洗管路,有利于仪器的稳定。
用HPLC测氨溴索含量时,片子有包衣,溶剂用0.1%HCL溶解.连续进针后就发现有杂质明显增大的情况.后改进为:用5ml0.1%HCL先溶样品,再用0.5%亚硫酸氢钠定容,就不会出现杂质增大的现象。
使用一段时间后会出现柱头塌陷,造成双头峰,可用同种牌号的填料将塌陷填平整,即可恢复分离效果,节约检验费用。 不过一般回填效果和使用时间也不会特别满意。
用液相色谱法测含量时,跑基线的时间一定要足够长,有时基线虽在短时间内平了,但
还没有充分饱和,导致在用了柱温箱的情况下,出峰的保留时间也会有很大差异!
进行HPLC检测使用磷酸盐缓冲液:乙腈做缓冲液时时,当缓冲液比例小于50%时,由于混合过程是吸热反应,在该过程中磷酸盐会析出晶体,造成流动相混浊而报废。如强行使用会阻塞色谱管道,对于这种情况,可以有以下两种处理方法,一时首先配制50:50的溶剂,然后10%加入,超声至高于室温,在加入10%,再超声高于室温,直至到达所需比例。另一种是首先配制不带盐的流动相,再超声至高于室温后,加入固体盐,超声至溶解,再过滤。
检验杂质时,如果流动相用的是缓冲盐类的,要定期冲洗液相系统,保证杂质的检验准确度。
HPLC应用醋酸水溶液作流动相时要注意作完后要长时间冲洗柱子,否则会堵住的。
1、刮掉表面黄色或褐色的填料,用新的同样的填料填补一下;
2、把过滤头用30%硝酸超声30分钟,用纯水超至中性,
在做HPLC的梯度洗脱检验时,除了柱温、流动相的PH值、两相比例的比例外,进样时的柱压也是影响出峰时间和形状的一个重要因素,所以进样前流动相的比例、运行时间及进样时的压力要尽量保持一致,才能使试验的重复性符合要求。
药物分析,所用的试剂质量很关键,做抗生素的聚合物含量时,经常在聚合物出峰的时间也出现倒峰,查找了所有仪器和溶剂,最后确定是一个化学试剂厂生产的磷酸二氢钠的原因。
在使用一些易挥发性的试剂(如甲醇\乙醇或三氯甲烷)作溶剂时,操作动作要迅速,否则测量结果会比真实值偏高。
HPLC如果流动相有缓冲盐时,更换流动相时务必先将缓冲盐更换掉,否则会将堵塞
。
分子筛
的使用:一般而言分子筛
不如反相
的寿命长,有时维护不好做上两百多个样品就不行了,所以每次使用完一定要充分地用超纯水把盐冲洗干净,并且用0.05%的叠氮钠保存,一般冲洗两小时就可以了。曾经也低速冲洗过夜的,后来发现可能是水对硅醇基的影响反而降低了柱子的寿命,就改了。可以在软件上设置自动关泵,就不用人一直看着啦。
做HPLC时,样品稀释好后是需要过滤的,最好选用与生产使用的同等材质的滤膜,这样就不会产生偏差了。
2、更换流动相后,瓶子的体积忘记修改,结果不是进气泡就是中途强制停止运行;
3、走序列时,忘记勾选shut down,以致到了早上出错报警;
4、缓冲液的pH一定要调节准确,否则对RT很有影响的;
用HPLC法测物质有关物质时,样品放置的时间以及放置的温度极其重要,所以最好现用现配,或者配好的样品液一定要低温保存,条件允许的话可以加一个自动上样控温装置,这样可防止杂质的降解,保证结果准确度。
走访过很多药厂,查看HPLC时大多数都可以在泵头和管路连接处发现有白色的结晶出现。产生这种结晶的原因是使用了含缓冲盐的流动相后,对系统冲洗时间不够,管路中残留了少量的缓冲盐随流动相渗出造成的。如果发现相同的问题,只要延长冲洗时间就可以解决。
做滴定时一定要注意所配溶液是否和以前的溶液的性状相同(如:颜色),如果不同就要考虑试剂、溶剂和器皿是不是变质或被污染了。
做紫外时因重视仪器的预热时间,预热时间太短,对实验结果的影响很大。
看到有很多实验员在配置薄层板的CMC-Na溶液时喜欢加热,使其快速完全的溶解。这样做有一个很大的弊端,制作出来的薄层板是非常不平滑的,建议还是让其自己慢慢溶解,即时不能完全溶解,也可以使用。
岛津的HPLC-10A的部分仪器对乙腈非常敏感,如果流动相中含有大量的乙腈时因逐步过度,否则会产生漏液或压力不稳定。7.做HPLC时使用低波长时,水的纯度要求要比高波长要高一些。比如210nm以下波长检测时。
HPLC中流动相中加了三乙胺可以减小拖尾,关键是PH值。
在做液相时候,如果保留时间不一致,看看室内温度变化情况,还有就是你要是手动进样时,进样速度也有关系!
在做不同厂家同一原料药的熔点时发现该原料药的熔点均不符合规定,在查、做液相时如果流动相有缓冲盐,一定要注意你的
的 冲洗。不然峰的分离度不好。
做药品有机溶剂残留量检查时,可以不拘泥于规定的
, 通常的DB-624可以满足要求, 取样量也可以灵活调整, 标准溶液浓度根据限度做相应调整就可以了。
在做有机溶剂残留时要注意载气流速一般柱流速在1-3ml较好,太大样品重复性不好,尾吹气流量也需注意。
用GC测有机残留水不溶性成分时,如苯、二氯甲烷等,称取毫克级标样时,在量瓶底部预先加少量DMF/DMSO,会减少天平的跳动,帮助准确称量。










加载更多