点击化学——释义与目标
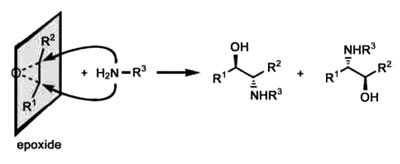
图1. 胺进攻的环氧化物开环反应: 一个典型的点击反应。
2001年,Scripps研究所的化学家、诺贝尔化学奖获得者K.Barry Sharpless提出点击化学(Click chemistry)概念,其主旨是开辟以碳-杂原子键(C-X-C)合成为基础的组合化学新方法,并借助点击反应来简单高效地获得分子多样性。点击化学对化学合成领域有很大的贡献,在药物开发和分子生物学的诸多领域中,它已经成为目前最为有用和吸引人的合成理念之一。
化学,是一种致力于使功能与形式紧密结合于其中的构造艺术尝试。正因为分子的功能是最重要的,从药物的发现到新塑料、香水、染料等的发明,创造新的功能一直是化学所有努力的目标。而可能的结构的范围和多样性是不可限量的,有人做过计算,仅仅9 种最常见的元素就可以组成1063种相对较小的分子,这个惊人的数字大概是太阳中原子数目的100万倍。
在这个浩瀚的潜在备选分子结构库中,肯定有着解决各种化学问题的答案,那就是,许多不同的分子有着人们期待的功能。而困难当然就是去找到这些分子。新功能分子的创造,往往是利用类似结构(做出与拥有目标功能的已知结构相类似的结构),或者从未经检验的结构中搜寻。即使是后者,化学家们还是倾向于停留在熟悉的区域。部分困难来自于我们现有的工具:当你拥有的仅是用于制造汽车的螺帽、螺丝和扳手的时候,你就很难制造出一辆与汽车区别很大的运输工具。正是这种连接,这种螺帽2螺丝具体细节的连接,决定了什么可以被组装起来和什么可以被制造出来。
#p#
在现代化学150 余年的历史中,发展出了将分子片段相互连接的多种技术。其中有相当多是很精致的,要求在严格控制的条件下细致地操作高活性的反应物。K.Barry Sharpless给那些最佳的化学反应起了一个名字 “点击化学”。这些反应易于操作,并能高产率生成目标产物,很少甚至没有副产物,在许多条件下运作良好(通常在水中特别好),而且不会受相连在一起的其他官能团影响。“点击”这个绰号意味着用这些方法把分子片段拼接起来就像将搭扣两部分“喀哒”扣起来一样简单。无论搭扣自身接着什么,只要搭扣的两部分碰在一起,它们就能相互结合起来。而且搭扣的两部分结构决定了它们只能和对方相互结合起来。
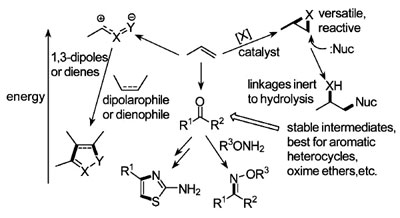
图2. 点击化学采用高能量,但带选择活性的官能团。
Sharpless及其同事提出来的中心问题就是,能否用这种最佳(“点击”)化学反应合成的分子来实现希望得到的功能。斟酌避免使用更复杂的合成技巧,对于通常使用复杂方法合成复杂结构的有机化学家来说,完全是一种挑战(能用简单的方法合成复杂的结构吗?而对于功能高分子的合成则是必不可少的,如果不使用符合点击化学标准的反应,这种合成就无法实现。
点击化学反应
点击反应有着下列的共同特征:
(1)许多反应的组件是衍生于烯烃和炔烃,这些都是石油裂化的产物。从能量与机理的角度,碳2碳多重键都可以成为点击化学反应的活性组件。
(2)绝大部分反应涉及碳2杂原子(主要是氮,氧,硫)键的形成。这与近年来重视碳2碳键形成的有机化学方向不同。
#p#
(3)点击反应是很强的放热反应,通过高能的反应物或稳定的产物都可以实现。
(4)点击反应一般是融合(fusion)过程(没有副产物)或缩合过程(产生的副产物为水)。
(5)很多点击反应不受水的负面影响,水的存在反而常常起到加速反应的作用。这些特征可在环氧化物与多种不同亲核试剂的开环反应中展现出来。如图1,因为环氧化物是一个张力很大的三元环,开环反应是一个非常有利的过程。然而开环需要在特定的条件下发生:亲核试剂仅能沿着C-O键的轴向进攻其中一个碳原子,这样的轨道排列不利于与开环反应竞争的消去反应,从而避免了副产物并得到高的产率。此外,环氧化物与水反应的活性不高,而水的形成氢键能力与极性本质都有利于环氧化物与其它亲核试剂进行开环反应。
大部分点击反应许多年前就已经发现并广泛应用了,但它们尚未被充分地利用。这包括以下的几类反应(见图2)。
A.张力环亲电试剂的亲核开环反应。环氧化物,吖丙啶,吖丙啶离子,环硫离子等三元环的开环反应,都是易于实现和多用途的反应。这一类反应还包括α,β-不饱和羰基化合物的迈克尔加成(Michael addition)反应。
B. 羰基化合物温和的缩合反应。这类可靠而广泛应用的反应包括(1)醛或酮与1,3-二醇反应生成1,3-环氧戊环;(2)醛与肼(hydrazines)或胲(hydroxylamine ethers)反应生成腙(hydrazones)和肟(oximes);(3)α-和β-羰基醛, 酮和酯生成杂环化合物。
C. 环加成反应。通常其反应基团是相对非极性的,这些融合反应涵盖广泛的反应,如Diels-Alder反应。最有用的是1,3-偶极环加成反应,其中以叠氮化物和炔的反应最为突出。
叠氮化物-炔环加成反应
点击化学的目标和思想,在非催化(图3A)及铜催化(图3B)的叠氮化物2炔环加成反应中体现得最充分。前者由A. Michael 于1893年第一次报道,并在20 世纪60年代至80年代Rolf Huisgen的研究中得以透彻理解并确立为一类重要的新反应。2001 年,丹麦的Meldal 研究组与美国的Sharpless研究组分别独立发现了铜催化反应。这一反应需要采用端基炔烃。其中用缩写AAC(azide-alkyne cycloaddition)代表非催化的过程,用CuAAC 代表铜催化的过程。
#p#
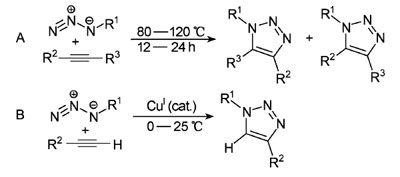
图3. 非催化(A)及铜催化(B)的叠氮化物-炔环加成反应。
由于其反应基团的特殊性质,这些反应非常有用。叠氮化物和炔烃的化学势能都很高(热力学不稳定),它们融合成三唑环时放出大于188千焦/摩的热量。而另一方面,这一反应的速率很慢,对于非活化(不是非常缺电子,也没有张力)炔烃,一般需要长时间加热。叠氮化物和炔烃对亲核试剂、亲电试剂和一般的溶剂均表现出惰性,目前,叠氮化物是唯一有此性质的1,3-偶极试剂。更重要的是,叠氮化物和炔烃几乎完全不与生物分子发生反应。它们小,不能形成强氢键,极性相对弱,对连接在其上的其他结构的性质没有显著的影响。而且,它们都可以很容易地引入到有机化合物中。
由于叠氮化物和炔烃的特殊活性:对其它所有试剂的惰性及相互反应的缓慢,它们可被利用于在酶这一“反应容器”中来组装那些能与酶紧密结合的分子,如图4所示。这一技术,被称作“原位点击化学”(“click chemistry in situ”),用叠氮化物和炔烃来标记那些能结合酶上相近位置的分子。如果这些被标记的分子能够同时与目标作用,而使得在某个合适的方向上叠氮化物和炔烃足够的靠近,三唑环就可以生成并把这两部分与酶结合的组件联结起来。因为双臂结合总是比单臂结合要强,于是就可以得到一个能结合得更紧密的分子。这一技术不需要事前了解目标酶的结构,也不需要对酶进行活性测试。因为在这些实验中,如果叠氮化物和炔烃标记的分子没有结合到酶模板的合适位置,溶液中叠氮化物和炔烃的浓度使之不足以发生反应,所以,这个可用质谱轻易探测的三唑环产物一旦生成,就证明一个极佳的酶抑制剂的诞生。
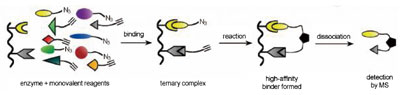
图4. “原位点击化学”技术。
#p#
原位点击化学(click chemistry in situ)已被用来发现多种酶的高亲和力的抑制剂,包括重要的神经递质酶(neurotransmitter enzyme),如乙酰胆碱酯酶(acetylcholinesterase);新陈代谢酶(metabolic enzyme),如碳酸酐酶(carbonic anhydrase);和艾滋病毒(HIV)蛋白酶(HIV protease)。在这些和其它的研究中,可以明显看到三唑环在药物开发中有着优越的特性。它有着大的偶极距,可以形成强的氢键,能够参与π2堆积作用,三唑环可以多种形色与蛋白发生作用。把两个“看不见的”组件在酶的空腔中合成三唑环这一发现正影响着原位药物开发工作中成键的选择性。原位点击化学技术,作为对传统药物合成与筛选方法的补充,正被世界上很多 和药物公司所采用。
由于铜的细胞毒性和伴随生理调节(attendant bioregulation),铜催化的反应还未能直接应用于活体细胞中,然而铜催化反应已在有机和材料科学中得到了格外广泛的应用。这些应用包括合成生物活性化合物,制备蛋白和聚核苷酸的共轭体(conjugates toproteins and polynucleotides),合成染料,对已知高分子的改进和合成新型高分子, 创造响应材料(responsive materials),以及在表面上以共价键连结目标结构。其在新药开发上的应用已有综述。这个反应正被深入研究,新的应用正在加速出现。
结论
点击化学是一种简单的合成方式,以实现和创造新功能物质和材料为目的。它在很大程度上已取得成功,并将得到持续的发展。然而,铜催化的三唑环合成只是目前最成功的例子,它远不是精华所在。不难理解,很多化学家认为点击化学仅仅是一个单一的反应。我们希望,随着时间的推移,学术和实践经验能打破这一视野的限制。
点击化学拓展着结构的领域,这些结构可以由专业化学家,也可以由非化学家合成出来。基本原理很简单:化合物片断的连结反应越能抵抗外界影响,就越会有多样的片断得以连结以解决各种问题。化学家没有像活细胞那样控制反应的能力,也没有制造如蛋白那样多功能的大结构分子的能力。所以,大自然可以启示我们,而我们需要耗费惊人的时间和费用来做出她的复杂分子。如果化学功能是我们的目的,那么,保持简单是一条值得记住的有益规则。
【参考文献】
[1] 2001年以来关于点击化学的文献,参见<http://www.scripps.edu/chem/sharpless/click.html>, 截至2007 年7 月,这一数字已超过900,其中铜催化三唑成环的反应占了近90 %。
展源
何发
相关文章
-
点击化学--释义与目标
2020-05-27
-
点击化学的释义与目标
2020-05-27
-

QC, IQC, IPQC, QA,到底是什么鬼?
2020-05-27
-

AAS法分析茶叶中的铅,镉,砷
2020-05-27
-
红外光谱分析,你了解多少?
2021-01-11
-

HPLC检测器,你了解吗?
2024-03-06
-

三聚氰胺,你还要害多少人
2020-05-27
-

选对 ,快速开发方法
2020-05-27
-

超净工作台原理,使用与维护
2020-05-27

加载更多